|
CONGENITAL ADRENAL
HYPERPLASIA |
The term congenital adrenal hyperplasia encompasses several autosomal recessive disorders that share complete or partial deficiency of an enzyme involved in cortisol or aldosterone synthesis. All disorders of this group share the common feature of a deficiency or relative defect in cortisol or aldosterone synthesis resulting in some degree of cortisol or aldosterone deficiency, or both. The affected enzyme can be totally or partially impaired. The degree of enzyme insufficiency determines the severity of the condition.2,5
Patients with mild congenital adrenal hyperplasia are frequently unable to mount sufficient stress responses to trauma and infection. Glucocorticoid precursors accumulate in these persons and are converted to androgenic steroids, causing shortened stature, early puberty, severe acne, and virilization and infertility in females.2,3,5,6 Mineralocorticoid synthesis can also be affected, resulting in electrolyte disturbances, hypotension and syncope.5,6
INCIDENCE |
·
In the
Nonclassic adrenal hyperplasia may
occur with a frequency ratio as high as
· Internationally: 11-Beta-hydroxylase deficiency is more commonly observed in persons of Moroccan or Iranian Jewish descent. Other forms of congenital adrenal hyperplasia are far less common.
The incidence of classic congenital adrenal hyperplasia is
especially high in
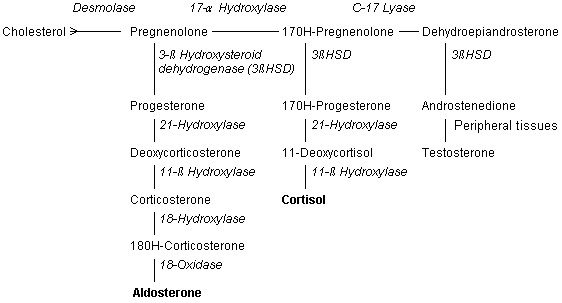
ENZYME PATHWAYS AND GENETICS |
|
Normal
enzyme pathway |
|
|
|
21-Hydroxylase |
|
Ninety percent of patients with congenital adrenal hyperplasia have 21-hydroxylase deficiency.2-4,6 Because this enzyme functions in both glucocorticoid and mineralocorticoid synthesis, some patients with 21-hydroxylase deficiency have insufficient amounts of cortisone and aldosterone). These persons have the "salt-wasting" form of congenital adrenal hyperplasia, with hyponatremia, hypovolemia, hyperkalemia and hypotension.1-4,6 The enzyme 21-hydroxylase is a chromosome 6, human leukocyte antigen (HLA)linked, cytochrome P450 enzyme that is found in the smooth endoplasmic reticulum. Its DNA sequence can be altered by at least nine mutations, many of which leave the enzyme impaired but not totally inactive.2,3,6 |
|
|

|
11-ß hydroxylase |
|
Deficiency of 11-ß hydroxylase is found in 8 to 9 percent of patients with congenital adrenal hyperplasia.2,5 Glucocorticoid synthesis remains impaired but, in this disorder, deoxycortisol accumulates. Deoxycortisol and its metabolites have mineralocorticoid properties and may cause hypertension when they accumulate.2,3,7 Thus, simple blood pressure measurements may help determine the underlying type of congenital adrenal hyperplasia. The enzyme 11-ß hydroxylase is a chromosome 8, cytochrome P450 enzyme located in the mitochondria. Known gene abnormalities include insertions, deletions, mis-sense/nonsense codons, and point mutations. Some of these abnormalities result in severe dysfunction of the enzyme while others result in only partial impairment.3-5 Classic 11-ß hydroxylase deficiency occurs in
approximately one per 100,000 births and occurs more frequently in Moroccan
Jews. Mild congenital adrenal hyperplasia due to 11-ß hydroxylase deficiency
is more common, however, and may be responsible for 1 to 2 percent of cases
of hirsutism and oligomenorrhea in women.3-5 |
|
|
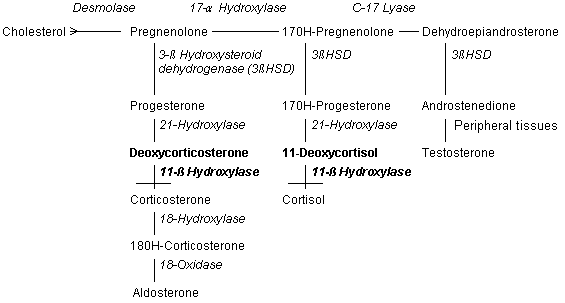
CLINICAL MANIFESTATIONS |
The clinical phenotype depends upon the nature and severity of the enzyme deficiency. 21-Hydroxylase deficiency (CYP21) is the most common form. Approximately 50% of patients with classic congenital adrenal hyperplasia from CYP21 deficiency have salt wasting due to inadequate aldosterone synthesis. Females with this disorder are usually recognized at birth because of ambiguous genitalia, but males have normal genitalia and are not diagnosed until later, often with a salt-wasting crisis. Females with less severe forms of 21-hydroxylase deficiency (ie, simple virilizing adrenal hyperplasia) are identified later in childhood because of precocious pubic hair and/or clitoromegaly, often accompanied by accelerated growth and skeletal development. Those individuals with mild deficiencies of the enzyme present in adolescence or adulthood with varying virilizing symptoms ranging from oligomenorrhea to hirsutism and infertility (nonclassic adrenal hyperplasia).
· Males with CYP21 deficiency are generally unrecognized at birth because their genitalia are normal. These individuals come to medical attention either at a few weeks of life with salt wasting resulting in dehydration, hypotension, hyponatremia, and hyperkalemia or later in childhood because of early pubic hair and/or phallic enlargement accompanied by accelerated linear growth and advancement of skeletal maturation.
· Males or females with CYP11 deficiency may present in early infancy (second or third week of life) with a salt loss crisis. This occurs in spite of the fact that these children usually develop hypertension or hypokalemic alkalosis, or both. This has been explained by the relative inability of the elevated 11-deoxycorticosterone levels to replace the defective levels of aldosterone, a phenomenon that disappears with the progression of age and the gradual decreased dependency of the infant from aldosterone.
· Infants with StAR deficiency (lipoid adrenal hyperplasia) usually have signs of adrenal insufficiency (eg, poor feeding, vomiting, dehydration, hypotension, hyponatremia, hyperkalemia). Some patients do not receive medical attention until late infancy. Males with this form of adrenal hyperplasia have female or ambiguous genitalia. Females have normal female genitalia. Curiously, females who survive do develop breasts and menstruate at puberty.
· Other forms of adrenal hyperplasia are characterized by disordered genital development in utero, lack of development of secondary sexual characteristics, or hypertension. For example, females with CYP17 deficiency are rarely identified at birth but come to medical attention later in life because of hypertension or failure to develop secondary sexual characteristics at puberty. Males with this disorder have ambiguous or female genitalia. In the latter case, individuals may be raised as females and come to medical attention later in life because of hypertension or lack of breast development.
· Patients with aldosterone deficiency of any etiology may have dehydration, hyponatremia, and hyperkalemia, especially with the stress of illness.
|
Signs and Symptoms Suggesting Mild Congenital Adrenal Hyperplasia |
|
Children |
|
• Moderate to
severe recurrent sinus or pulmonary infections
|
|
Adults |
|
• Childhood history
as defined above |
|
Women |
|
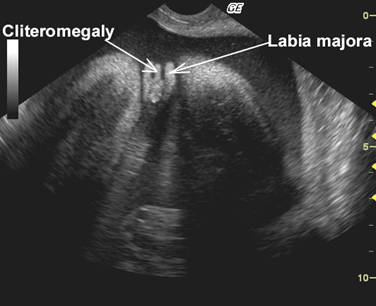
•
Clitorimegaly |
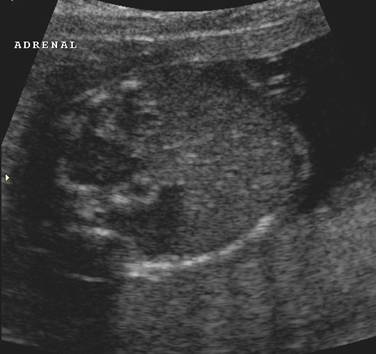
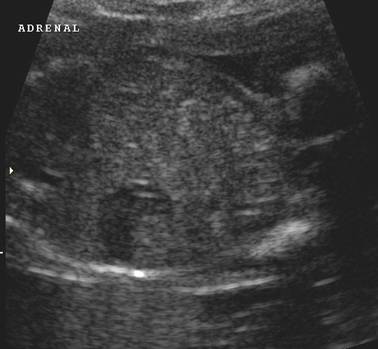
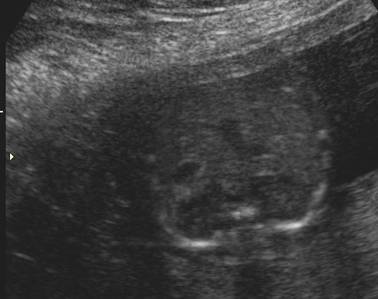
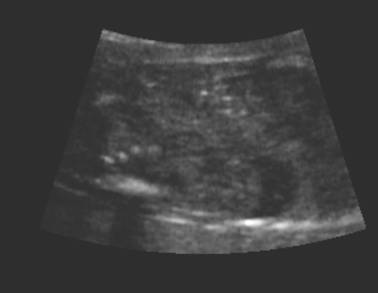
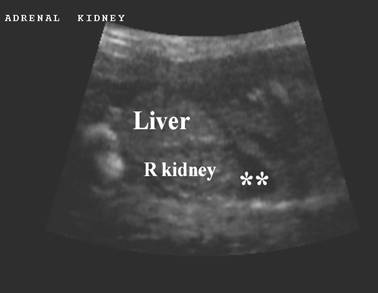
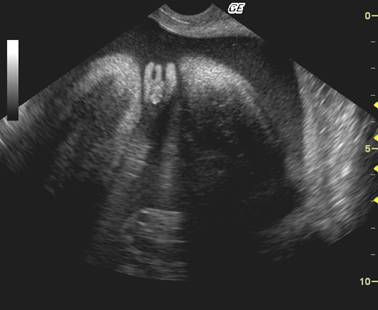
ULTRASOUND |
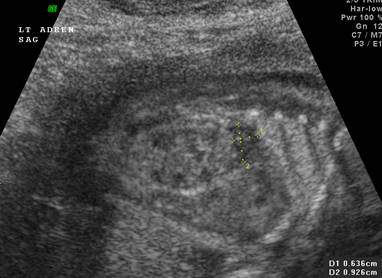
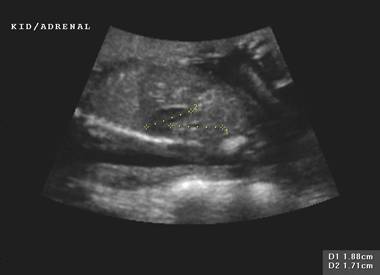
Normal adrenal gland – width of adrenal limb <4mm
- smooth surface
- echogenic central stripe with surrounding hypoechoic rim
Abnormal adrenal gland – increased in size
- lobulated cerebriform surface (wrinkled surface of the enlarged gland that suggests the appearance of brain gyri that is characteristic of CAH on pathologic examination).
- abnormal echogenicity
2 out of 3 abnormalities describes the diagnosis with a high degree of certainty (18).
These changes are reversible with the introduction of steroid replacement therapy.
Other findings include - Ambiguous genitalia (cliteromegaly n the female fetus). Virilization of the female fetus (treatment should begin before the development of the urogrnital sinus – 6-7 week of gestation – to prevent or decrease virilization (19).
Male fetuses – secretion of excessive androgendoes not appreciable affect male sexual differentiation (20).
|
Bilateral
adrenal enlargement |
|
|
|
|
|
|
|
|
3D volume image |
|
|
|
|
|
Cliteromegaly |
|
|
|
|
|
Follow
up 6 weeks after the mother was given oral steroid supplements |
|
|
|
|
DIFFERENTIAL DIAGNOSIS |
Anomalies that cause ambiguous genitalia: partial androgen insensitivity syndrome, maternal androgen ingestion or an androgen-secreting tumor during pregnancy.
REFERENCES |
- Theodoropoulou M, Barta C, Szoke M, Guttman A, Staub M, Niederland
T, Solyom J,Fekete G, Sasvari-Szekely M.Prenatal diagnosis of steroid
21-hydroxylase deficiency by allele-specific amplification.Fetal
DiagnTher2001Jul-Aug;16(4):237-40.
- Lee HH, Kuo JM, Chao HT, Lee YJ, Chang JG, Tsai CH,
Chung BC.Carrier analysis and prenatal diagnosis of congenital adrenal hyperplasia causedby
21-hydroxylase deficiency in Chinese.J Clin Endocrinol Metab 2000
Feb;85(2):597-600
- Lee HH, Kuo JM, Chao HT, Lee YJ, Chang JG, Tsai
CH, Chung BC.Carrier analysis and prenatal diagnosis of congenital adrenal
hyperplasia causedby 21-hydroxylase deficiency in Chinese.J Clin
Endocrinol Metab 2000 Feb;85(2):597- 600.
- Buyse, ML. Birth Defects Enciclopedia. Pag 1603
- Ritzen EM, Lajic S, Wedell A.How can molecular
biology contribute to the management of congenital adrenalhyperplasia?Horm
Res 2000;53 Suppl 1:34-7
- Pineda P, Fardella C, Poggi H, Torrealba I,
Cattani A, Soto J, Foradori A. Molecular diagnosis of salt wasting
congenital adrenal hyperplasia, caused by deficit of 21-hydroxylase, in
the Chilean population.Rev Med Chil 1997 Sep;125(9):987-92
- Kajic S, Bui TH, Holst M, Ritzen M, Wedell
A.Prenatal diagnosis and treatment of adrenogenital syndrome.
Preventvirilization of female fetuses.Lakartidningen 1997 Dec 10;94(50):4781-6
- Mathur R,
Kabra M, Menon PS. Prenatal diagnosis and treatment of steroid
21-hydroxylase deficiency (congenital adrenal hyperplasia).Indian J
Pediatr 2000 Nov;67(11):813-8
- Klein VR, Willman SP, Carr BR.Familial posterior
labial fusion.Obstet Gynecol 1989 Mar;73(3 Pt 2):500-3
- Mathur R, Kabra M, Menon PS.Diagnosis and
management of congenital adrenal hyperplasia: clinical, molecular and
prenatal aspects. Natl Med J India 2001 Jan-Feb;14(1):26-31
- Ko TM, Kao CH, Ho HN, Tseng LH, Hwa HL, Hsu PM,
Chuang SM, Lee TY. Congenital adrenal hyperplasia. Molecular
characterization. Reprod Med 1998 Apr;43(4):379-86
- Day DJ, Speiser PW, Schulze E, Bettendorf M,
Fitness J, Barany F, White PC.Identification of non-amplifying CYP21 genes
when using PCR-based diagnosis of 21-hydroxylase deficiency in congenital
adrenal hyperplasia (CAH) affected pedigrees.Hum Mol Genet 1996
Dec;5(12):2039-48
- Wedell A. Molecular approaches for the diagnosis
of 21-hydroxylase deficiency and congenital adrenal hyperplasia. Clin Lab
Med 1996 Mar;16(1):125-37
- VR, Willman SP, Carr BR.Familial posterior labial
fusion.Obstet Gynecol 1989 Mar;73(3 Pt 2):500-3
- Masturzo B, Hyett JA, Kalache KD, Rumsby G,
Jauniaux E, Rodeck CH. Increased nuchal translucency as a prenatal
manifestation of congenital adrenal hyperplasia.Prenat Diagn 2001
Apr;21(4):314-6
- Lajic S, Wedell A, Bui TH, Ritzen EM, Holst
M.Long-term somatic follow-up of prenatally treated children with
congenitaladrenal hyperplasia.J Clin Endocrinol Metab 1998
Nov;83(11):3872-80
- Mercado AB, Wilson RC, Cheng KC, Wei JQ, New MI. Prenatal treatment and diagnosis of congenital adrenal hyperplasia owing to steroid 21-hydroxylase deficiencyJ Clin Endocrinol Metab 1995 Jul;80(7):2014-20
- Al-Alwan I, Navarro O, Daneman D, Daneman A. Clinical utility of adrenal ultrasonography in the diagnosis of congenital adrenal hyperplasia. J Pediatr 1999;135:71-75.
- Chambrier ED, Heinrichs C, Avni FE. Sonographic appearance of congenital adrenal hypepplasia in utero. J Ultrasound Med 2002;21:97-100.
- White PC, Speiser PW. Congenital adrenal hyperplasia deficiency. Endocr Rev 200;21:245-291.