|
MEGACYSTIS-MICROCOLON-INTESTINAL HYPOPERISTALSIS
SYNDROME (MMIH) |
This is a rare congenital complex originally described in 1976 (1). It
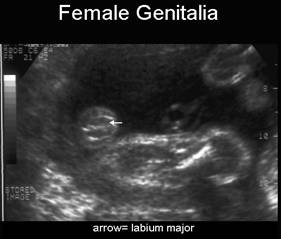
occurs more commonly in females (91%) although some males cases have been reported
(2,3). Most cases are sporadic,
however an autosomal recessive mode of inheritance
has been suggested in some reports of affected siblings (3-5). Suggested
etiology is an intramural inflammatory process affecting the gastrointestinal
and urinary tracts leading to extensive fibrosis. The fibrosis destroys the
intestinal neural network resulting in hypoperistalsis
and neuromuscular incoordination of the bladder. At
present it is still not clear whether this is a primary neuropathy or myopathy (6).
ULTRASOUND |
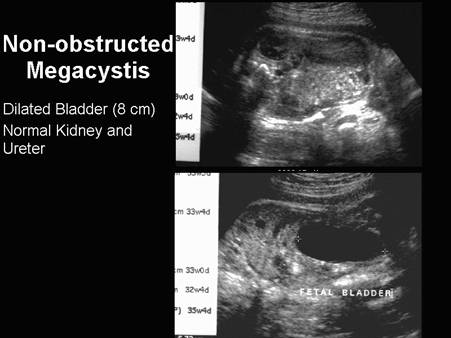
- Dilated, thick-walled, non-obstructed bladder (2,3).
- Bilateral hydronephrosis (7), but the ureters and kidneys may not be dilated (see case below) .
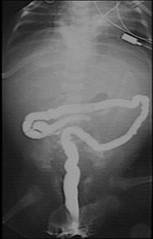
- Bowel is significantly shortened with dilatation of the proximal small intestines, a malrotated microcolon, and ineffective peristalsis producing a functional obstruction (3,7,8).
- Amniotic fluid volume is usually normal.
- Polyhydramnios has been reported (8,9).
- No hydroureters have been reported.
|
MMIH Syndrome
|
|
|
|
|
|
|
|
- Other less common reported findings include distended stomach, hydrometrocolpos, segmental colonic dilatation, omphaloceles and rhabdomyomata.
- Short bowel, microcolon and microileum cannot be antenatally detected.
DIFFERENTIAL DIAGNOSIS |
- Posterior urethral valves or urethral atresia (male fetuses without dilated ureters and oligohydramnios is present).
- In females, lower urinary tract obstruction (urethral stenosis / atresia) may produce similar genitourinary findings, however the oligohydramnios that would be present in these entities are not a feature of MMIH syndrome.
PROGNOSIS |
With few exceptions this syndrome is rapidly fatal (6% survivors at one year of age) (10). Despite hyperalimentation, intestinal function is usually the cause of death. Septicemia has been reported to cause the death of some infants (11).
Recurrence risk is 25% in cases of autosomal
recessive inheritance. Most cases are however sporadic.
REFERENCES |
- Berdon
WE, Baker DH,
- Krook PM. Megacystis-microcolon-intestinal hypoperistalsis syndrome in a male infant. Radiology 1980;136:649-650.
- Olivera G, Boechat MI, Ferreira MA. Megacystis microcolon intestinal hypoperistalsis syndrome in a newborn girl whose brother had prune belly syndrome: common pathogenesis? Pediatr Radiol 1983;13:294-296.
- McNamara HM, Onwude JL,
- Puri
P,
- Srikanth MS, Ford EG, Isaacs H Jr et.al. Megacystis microcolon intestinal hypoperistalsis syndrome: late sequelae and possible pathogenesis. J Pediatr Surg 1993;28(7):957-959.
- Jona JZ, Werlin SL. The megacystis-microcolon-intestinal hypoperistalsis syndrome: Report of a case. J Pediatr Surg 1981;16:749-751.
- Vezina WC, Morin FR, Winsberg F. Megacystis-microcolon-intestinal hypoperistalsis syndrome: Antenatal ultrasound appearance. Am J Roentgenol 1979;133:749-750.
- Manco L, Osterdahl P. The antenatal sonographic features of Megacystis-microcolon-intestinal hypoperistalsis syndrome. J Clin Ultrasound 1984;12:595-598.
- Anneren G, Meurling S, Olsen L. Megacystis microcolon intestinal hypoperistalsis syndrome (MMIHS), an autosomal recessive disorder: clinical reports and review of the literature. Am J Med Genet 1991;41(2):251-254.
- Yokoyama S, Fujimoto T, Tokuda Y et.al. Successful nutrition management of megacystis-microcolon-intestinal hypoperistalsis syndrome – a case report. Nutrition 1989;5(6):423-426.