|
CHONDRODYSPLASIA PUNCTATA (1-3) |
Chondrodysplasia punctata refers to a heterogeneous group of conditions which share craniofacial dysmorphism and joint contactures and can be either rhizomelic, mesomelic or both. There is also calcific stippling of cartilage and periarticular soft tissues and, in particular, punctate calcification in the heel, in infancy. These disorders differ in clinical features, severity, inheritance pattern and radiological features, and an agreed upon classification has yet to be established.
|
Classification of chondrodysplasia
punctata |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Post natal XRay |
|
|
|
|
|
|
|
|
|
|
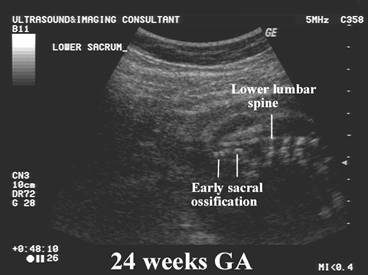
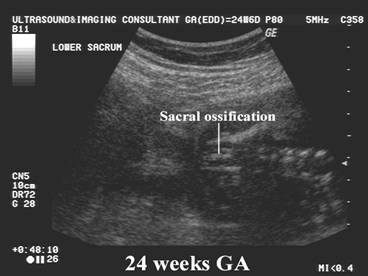
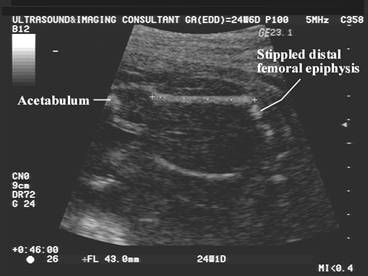
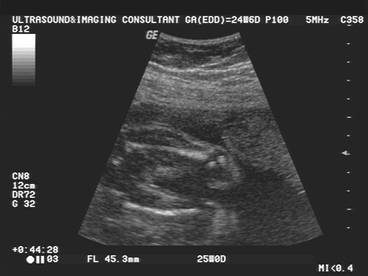
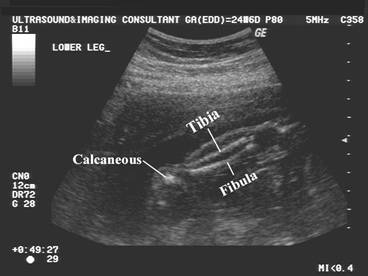
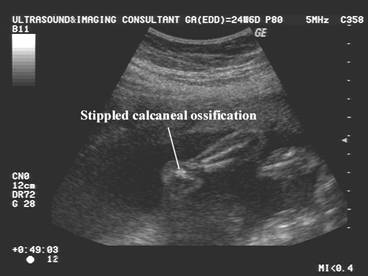
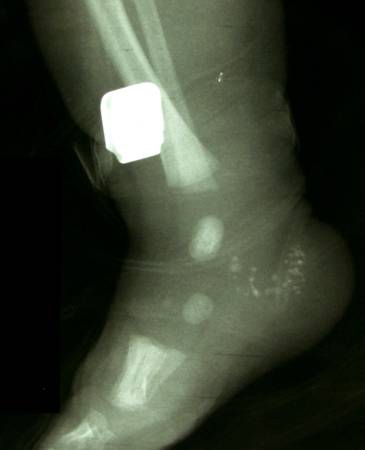
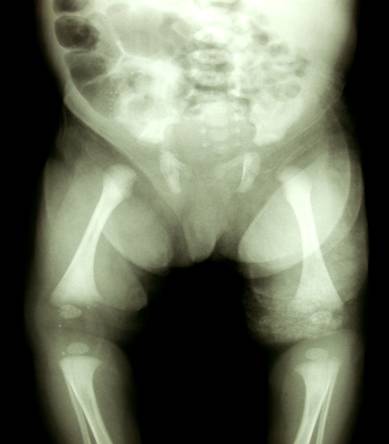
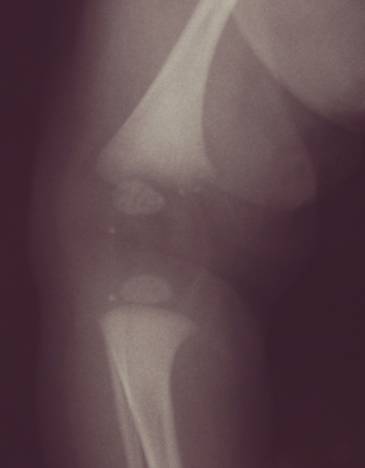
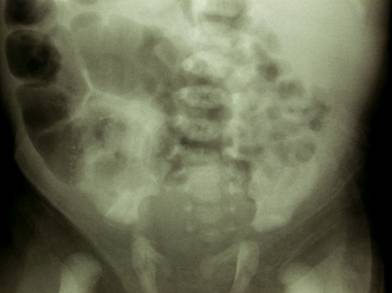
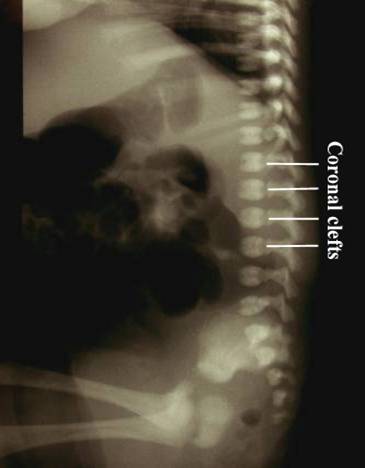
|
|
Video clip of Chondodysplasia
Punctata
|
|
|
|
|
|

DIFFERENTIAL DIAGNOSIS |
REFERENCES |
- Jansen V, Sarafoglu K, Rebarber A et.al. Chondrodysplasia punctata, tibial-metaphyseal type in a 16 week fetus. J Ultrasound Med 2000;19:719-722.
- Matsui M, Honma Y, Oguro N
et.al. Case report: A newborn case of chondrodysplasia punctata,
tibial-metacarpal type. Br J Radiol 1994;67:97.
- International Working Group on Constitutional Disease of Bone: International classification of osteochondrodysplasias. Am J Med Genet 1992;44:223.
- Bennett CP,
Am J Med Genet 1992;44:795-799. - Borochowitz Z. Generalized chondrodysplasia punctata with shortness of humeri and brachymetacarpy: humero-metacarpal (HM) type: variation or heterogeneity? Am J Med Genet 1991;41:417-422.
- Argo KM, Toriello HV,
Jelsema RD, Zuidema LJ.
Prenatal findings in chondrodysplasia punctata, tibia-metacarpal type. Ultrasound Obstet Gynecol 1996;8:350-354. - Rittler M, Menger H, and Spranger J. Chondrodysplasia punctate, tibia-metacarpal (MT) type. Am J Med Genet 1990;37:200-208.
- Braverman N, Steel G, Obie
C, Moser A, Moser H, Gould SJ, Valle D.
Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata.
Nat Genet 1997;15:369-376. - Bruch D, Megahed M,
Majewski F, Ruzicka T.
Ichthyotic and psoriasiform skin lesions along Blaschko's lines in a woman with X-linked dominant chondrodysplasia punctata.
J Am Acad Dermatol 1995;33:356-360. - Fourie DT. Chondrodysplasia
punctata: case report and literature review of patients with heart
lesions.
Pediatr Cardiol 1995;16:247-250.
Fryburg JS, Kelly TE. Chondrodysplasia punctata, humero-metacarpal type: a second case. Am J Med Genet 1996;64:493-496 - Gobello T, Mazzanti C,
Fileccia P, Didona B, Papi M, Atzori F, Cavalieri R. X-linked dominant
chondrodysplasia punctata (Happle syndrome) with uncommon symmetrical
shortening of the tubular bones.
Dermatology 1995;191:323-327. - Gobello T, Mazzanti C,
Fileccia P, Didona B, Papi M, Atzori F, Cavalieri R.
X-linked dominant chondrodysplasia punctata (Happle syndrome) with uncommon symmetrical shortening of the tubular bones.
Dermatology 1995;191:323-327. - Gobello T, Mazzanti C,
Fileccia P, Didona B, Papi M, Atzori F, Cavalieri R.
X-linked dominant chondrodysplasia punctata (Happle syndrome) with uncommon symmetrical shortening of the tubular bones.
Dermatology 1995;191:323-327.